This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
What is Phylogenetics?
Phylogenetics is the study of evolutionary relationships among biological entities - often species, individuals or genes - which are often referred to as taxa. A phylogenetic tree can be constructed by looking at the nucleotide or protein sequences and combining this with an understanding of sequence evolution, which is described using an evolutionary model. This enables us to infer evolutionary events that happened in the past, and also provides more information about the evolutionary processes operating on sequences. Thus we may refine our understanding of how evolution works [1].
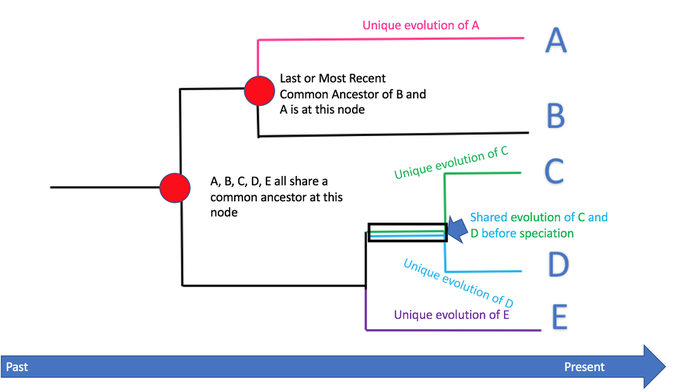
Figure 1. Phylogenetic tree diagram.
Methods For Constructing Phylogenetic Trees [2]
Maximum Likelihood
In this method, an initial tree is built using a method such as Neighbor Joining. The branch lengths of that tree are then altered to maximize the likelihood of the data set for that tree topology under the desired model of evolution.
In this method, an initial tree is built using a method such as Neighbor Joining. The branch lengths of that tree are then altered to maximize the likelihood of the data set for that tree topology under the desired model of evolution.
Neighbor Joining
The Neighbour Joining method is a method for re-constructing phylogenetic trees, and computing the lengths of the branches of this tree. In each stage, the two nearest nodes of the tree (the term "nearest nodes" will be defined in the following paragraphs) are chosen and defined as neighbours in our tree. This is done recursively until all of the nodes are paired together. Neighbor Joining employs a distance matrix, which uses physical separation between homologs to represent evolutionary separation. Uniquely, this method allows for unequal rates of evolution in different branches of the tree.
The Neighbour Joining method is a method for re-constructing phylogenetic trees, and computing the lengths of the branches of this tree. In each stage, the two nearest nodes of the tree (the term "nearest nodes" will be defined in the following paragraphs) are chosen and defined as neighbours in our tree. This is done recursively until all of the nodes are paired together. Neighbor Joining employs a distance matrix, which uses physical separation between homologs to represent evolutionary separation. Uniquely, this method allows for unequal rates of evolution in different branches of the tree.
Average Distance
Average distance takes scores generated by sequence similarities and joins the identical species together with equal branch lengths, assuming species diverged equally from a common ancestor.
Average distance takes scores generated by sequence similarities and joins the identical species together with equal branch lengths, assuming species diverged equally from a common ancestor.
NRXN3 Phylogenetic Tree Construction
Compiling Sequences
To begin a phylogenetic analysis, you must first compile protein sequences of NRXN3 homologs in various organisms. This can be achieved with GenBank and utilizing the Homologene tool. The sequences are then placed into a plain text file in a FASTA format. The slideshow below shows the FASTA format for the protein sequences of a select few organisms.
To begin a phylogenetic analysis, you must first compile protein sequences of NRXN3 homologs in various organisms. This can be achieved with GenBank and utilizing the Homologene tool. The sequences are then placed into a plain text file in a FASTA format. The slideshow below shows the FASTA format for the protein sequences of a select few organisms.
Aligning Sequences
The compiled sequences can then be aligned using ClustalWOmega.
The compiled sequences can then be aligned using ClustalWOmega.
Tree Construction
The aligned sequences can now be used to construct a phylogenetic tree on ClustalWOmega like the one shown below.
The aligned sequences can now be used to construct a phylogenetic tree on ClustalWOmega like the one shown below.
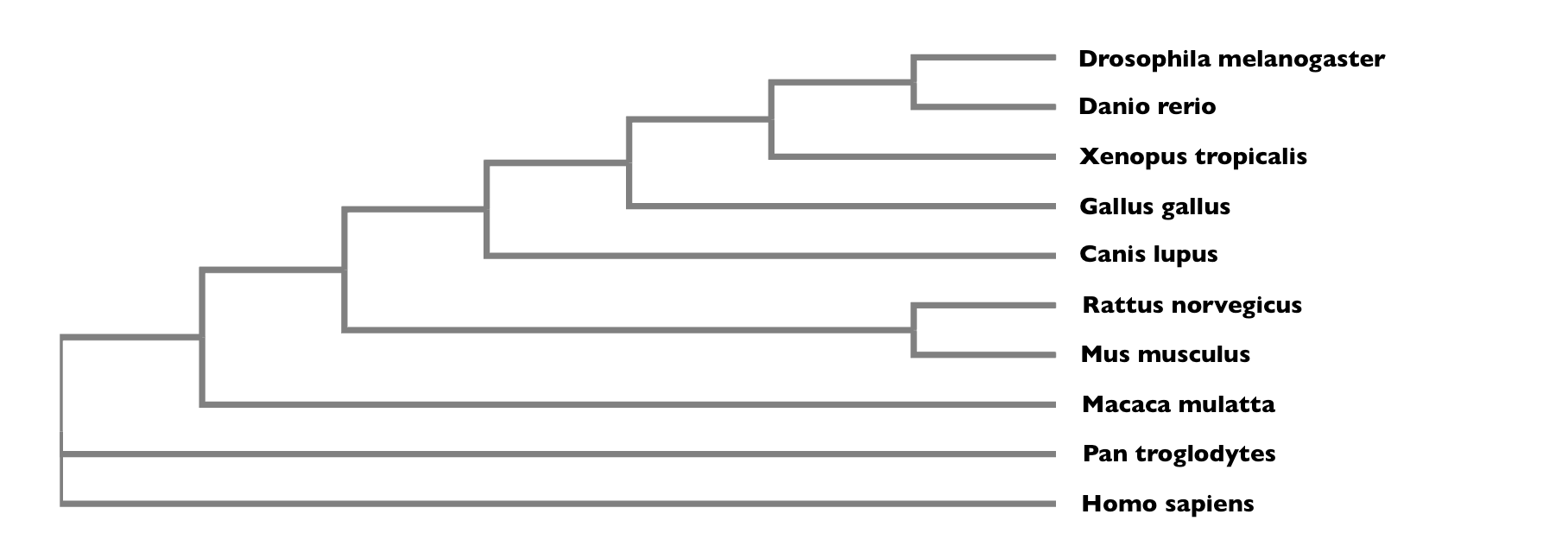
Neighbor Joining Tree
Conclusion
Protein phylogenetics allows us to analyze the conservation and presence of NRXN3 throughout different homologous organisms. The percent identities that were found for each homolog through BLAST are verified through protein phylogenetics. By creating a phylogenetic tree, we can see how conserved NRXN3 is across all homologous organisms. Even though percent identities are not always set in stone and do not always reflect how conserved a gene is in an organism, the phylogenetic tree constructed above does agree with the percent identities that I found via reciprocal BLASTs. The percent identities for for Drosophila melanogaster (35%) and Danio rerio (81%) were the lowest among all the homologs I had chosen. This is reflected in the tree above because both species are the furthest away from the root of the tree where Homo sapiens and other species with high percent identities were located. The phylogenetic tree then gives us information about the evolutionary patterns of each species and the NRXN3 protein.
References
[1] What is phylogenetics? (2016, June 08). Retrieved from https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
[2] Chhotwani, M., Francis, S., & Pal, A. (n.d.). Comparison of different phylogenetic tree generation methods. Retrieved from https://cise.ufl.edu/~sarath/BioInformatics/
Images
Header: https://macinivnw.deviantart.com/art/forest-trees-nature-green-wood-sunlight-backgroun-377394117
https://d2gne97vdumgn3.cloudfront.net/api/file/BFTdlb0TtGGboJByHyAh
[1] What is phylogenetics? (2016, June 08). Retrieved from https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
[2] Chhotwani, M., Francis, S., & Pal, A. (n.d.). Comparison of different phylogenetic tree generation methods. Retrieved from https://cise.ufl.edu/~sarath/BioInformatics/
Images
Header: https://macinivnw.deviantart.com/art/forest-trees-nature-green-wood-sunlight-backgroun-377394117
https://d2gne97vdumgn3.cloudfront.net/api/file/BFTdlb0TtGGboJByHyAh
